

Auteur: Benjamin Neveu, biochimiste clinique, DEPD, CSPQ, Institut de cardiologie de Montréal, Laboratoire de diagnostic moléculaire
Le séquençage parallèle à haut débit (SPHD) est un outil maintenant bien implanté dans les laboratoires diagnostiques. Cette technologie de pointe permet d’évaluer en simultané diverses altérations génétiques pouvant être d’intérêt clinique tant au niveau diagnostic, pronostique et thérapeutique. En fonction des bases pathophysiologiques des maladies investiguées, plusieurs alternatives de séquençage peuvent être employées afin de maximiser l’utilité clinique du test génétique tout en étant respectueux des ressources du Réseau de la Santé. En ce sens, des analyses ciblées (quelques dizaines de gènes) peuvent être indiquées pour certaines maladies héréditaires cardiovasculaires, alors que des analyses plus exhaustives (séquençage de l’exome ou du génome entier en trio) seront nécessaires afin de mettre en évidence l’altération génétique responsable d’un retard global de développement chez l’enfant, par exemple.
Peu importe l’étendue que prendra le séquençage, il se doit d’être en mesure de détecter à la fois des substitutions de nucléotides (SNV), de courtes insertions ou délétions (InDel; généralement moins de 50 paires de bases), ainsi que des changements du nombre de copies (Copy Number Variant; CNV) (Figure 1).
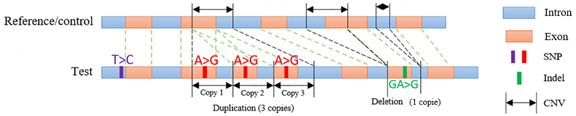
Figure 1: Les divers types de variations génétiques détectables en SPHD. Adaptée de : PMID 32153642, 2020.
Les changements structuraux (p.ex. inversions, translocations) sont exclus de la portée du SPHD et d’autres outils de diagnostic moléculaire plus adaptés sont disponibles afin d’en faire la recherche spécifique. Bien que les CNV soient des événements très rares, voir même anecdotiques dans certaines conditions, il n’en demeure pas moins essentiel d’en faire l’identification car ceux-ci peuvent être d’une grande utilité clinique.
La stratégie derrière l’appel de CNV par SPHD
Comme leur nom l’indique, les changements du nombre de copies sont un gain ou une perte de matériel génétique généralement définis comme ayant une taille supérieure à celle d’un exon. Contrairement aux SNV et aux InDel identifiés dans l’analyse bio-informatique comme étant les différences obtenues suite à l’alignement des séquences générées (reads) sur le génome de référence (Figure 2), l’analyse des CNV fait appel à une quantification basée sur la profondeur de couverture, c’est-à-dire le nombre de reads qui s’alignent à une même position (Figure 3). Ce n’est donc pas la séquence telle quelle qui est évaluée, mais bien le nombre de séquences alignées à un locus donné qui est à la source de leur identification. Bien qu’un seul et même essai de séquençage soit suffisant pour la génération des données nécessaires à l’identification de l’ensemble de ces altérations génétiques, cette distinction fondamentale explique les différences dans les processus analytiques et bio-informatiques subséquents nécessaires à l’identification différentielle des SNV/InDel et des CNV.
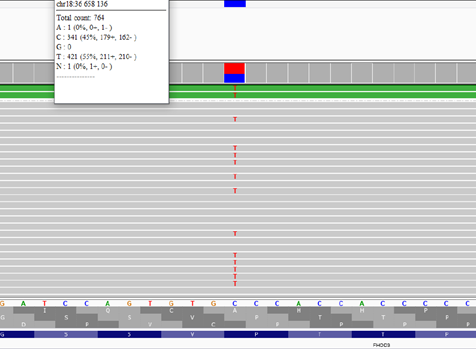
Figure 2 : Exemple de SNV. Visualisation d’une substitution nucléotidique (C>T) mise en évidence suite à l’alignement des séquences générées dans un essai SPHD. Chaque ligne fine représente une lecture de séquençage (read). En gris, les nucléotides séquencés s’alignent au génome de référence (indiqué par la suite de nucléotides dans le bas de la figure), alors que la base T (en rouge) est indiquée comme étant discordante avec la référence (base C attendue à cette position). La proportion (45% / 55%) des deux bases à cette position est compatible avec un SNV hétérozygote (i.e. un seul des deux allèles est porteur de cette substitution).
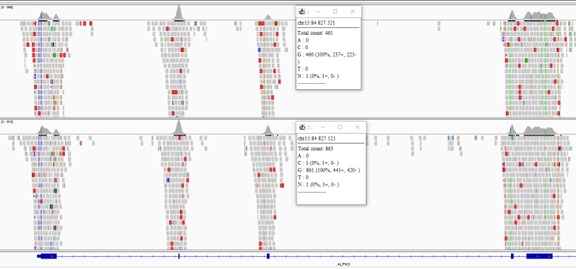
Figure 3 : Exemple de CNV. Visualisation d’un changement du nombre de copie à l’échelle d’un gène. Dans cet exemple, le séquençage ciblé génère un profil où seuls les exons (segments pleins sur la dernière ligne de la figure) sont couverts. Les introns ne sont pas ciblés et n’ont donc pas de couverture. En comparaison avec un échantillon référence (en bas), le spécimen en évaluation (en haut) affiche une profondeur de couverture (représentée par les courbes d’allure gaussienne) d’environ 50% celle de la référence, compatible avec une large délétion hétérozygote ciblée à l’exon 3 du gène ALPK3. Les exons 2 et 4 ont une profondeur de couverture similaire entre les deux spécimens, permettant de restreindre la délétion à l’exon 3.
Comme chaque série de séquençage est empreinte d’une certaine variabilité, il n’est pas recommandé d’évaluer les changements de profondeur de couverture sur la base de différences absolues. De plus, certaines régions sont systématiquement mieux couvertes que d’autres lors de l’emploi d’une approche de séquençage ciblé (certains gènes spécifiques ou l’exome), de sorte qu’une valeur seuil unique en termes de différence absolue du nombre de reads n’est pas acceptable pour la détection de CNV. Afin de mettre en évidence les CNV, une analyse comparative entre les profondeurs de couverture du spécimen en évaluation et celles d’un groupe contrôle (panel of normals) est nécessaire. Ainsi, la profondeur de couverture normalisée de chaque région (habituellement un seul exon) est comparée à celle obtenue chez des échantillons normaux (sans CNV).
Une approche classiquement employée comprend la génération d’un groupe contrôle qui utilise les autres spécimens de la même série analytique de séquençage. Comme plusieurs spécimens sont séquencés en parallèle (multiplexés, donc dans les mêmes conditions), ceux-ci peuvent être combinés et utilisés comme groupe contrôle, en assumant qu’ils soient exempts de CNV (comme les CNV sont des évènements excessivement rares, ceci est acceptable). Par exemple, si 48 spécimens sont multiplexés et séquencés sur la même flowcell, chaque spécimen est comparé aux 47 autres. Cette approche est relativement rapide et simple à mettre en place. De plus, si un biais systématique propre à cette série survient, il sera « normalisé » et grandement atténué. Toutefois, le nombre de spécimens servant de référence est limité par la capacité à multiplexer, et si une disparité, une anomalie ou un problème significatif survient dans une proportion significative des échantillons (p.ex. une préparation de librairie complète de 12 spécimens ayant une performance sous-optimale dans la capture), ceci aura une répercussion majeure sur la composition du groupe contrôle et pourrait provoquer une baisse de performance importante dans l’identification des CNV pour l’ensemble des spécimens de cette flowcell, jusqu’à la rendre impraticable. Pour que cette approche soit efficace et viable fonctionnellement, tous les spécimens de la série doivent donc être relativement homogènes. Même avec des processus robustes, les variations normales dans la qualité des ADN et des différents lots de réactifs peuvent altérer l’efficacité de cette approche pour la détection de CNV.
Ceci dit, des solutions à ces limitations existent! Une approche alternative innovante a été développée, validée et fera l’objet d’une autre publication : L’identification des changements du nombre de copies : un défi analytique et bio-informatique – Partie II.